This project presents a novel spatial proteomics workflow that maps protein expression at pseudo-single-cell resolution in human pancreatic islet tissue.
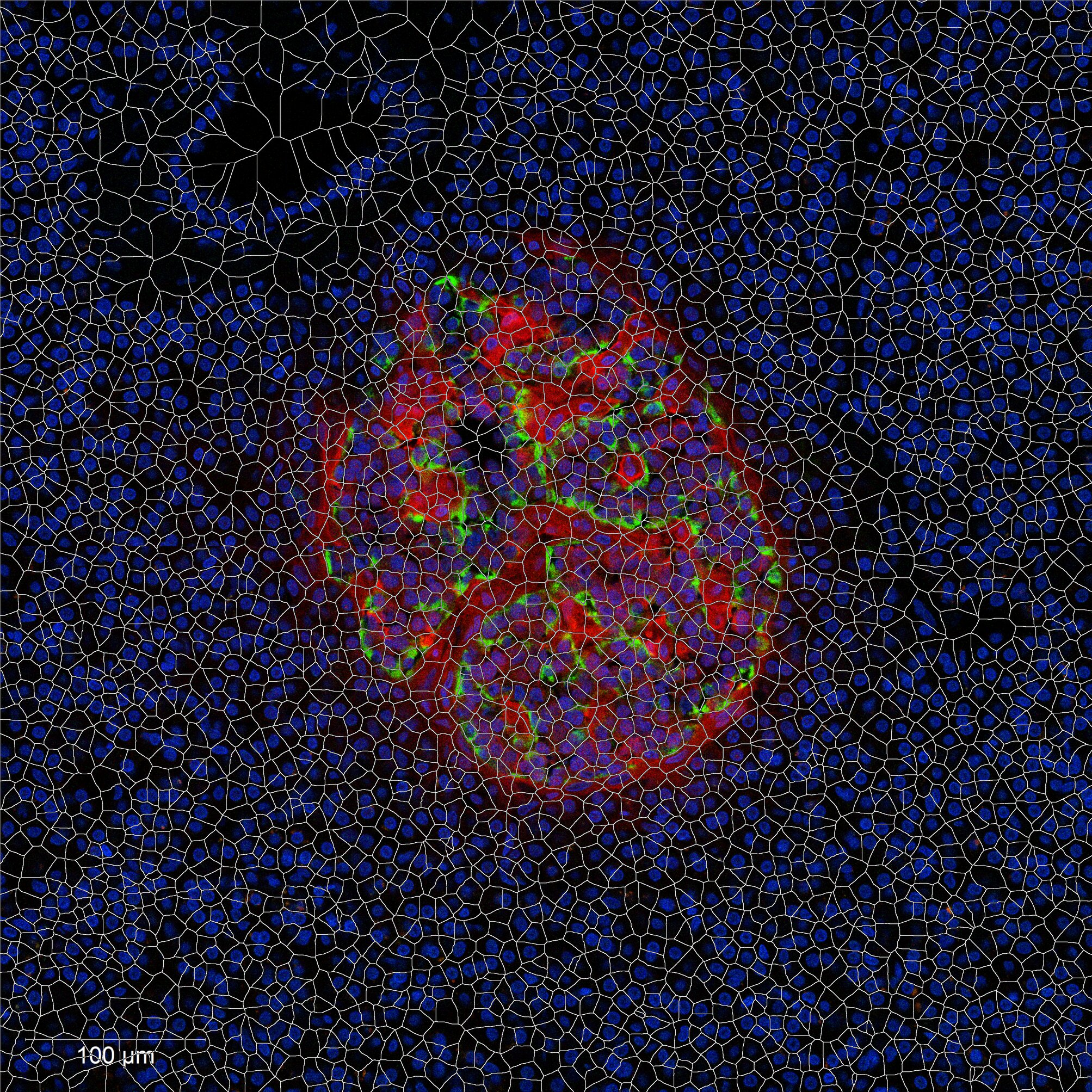
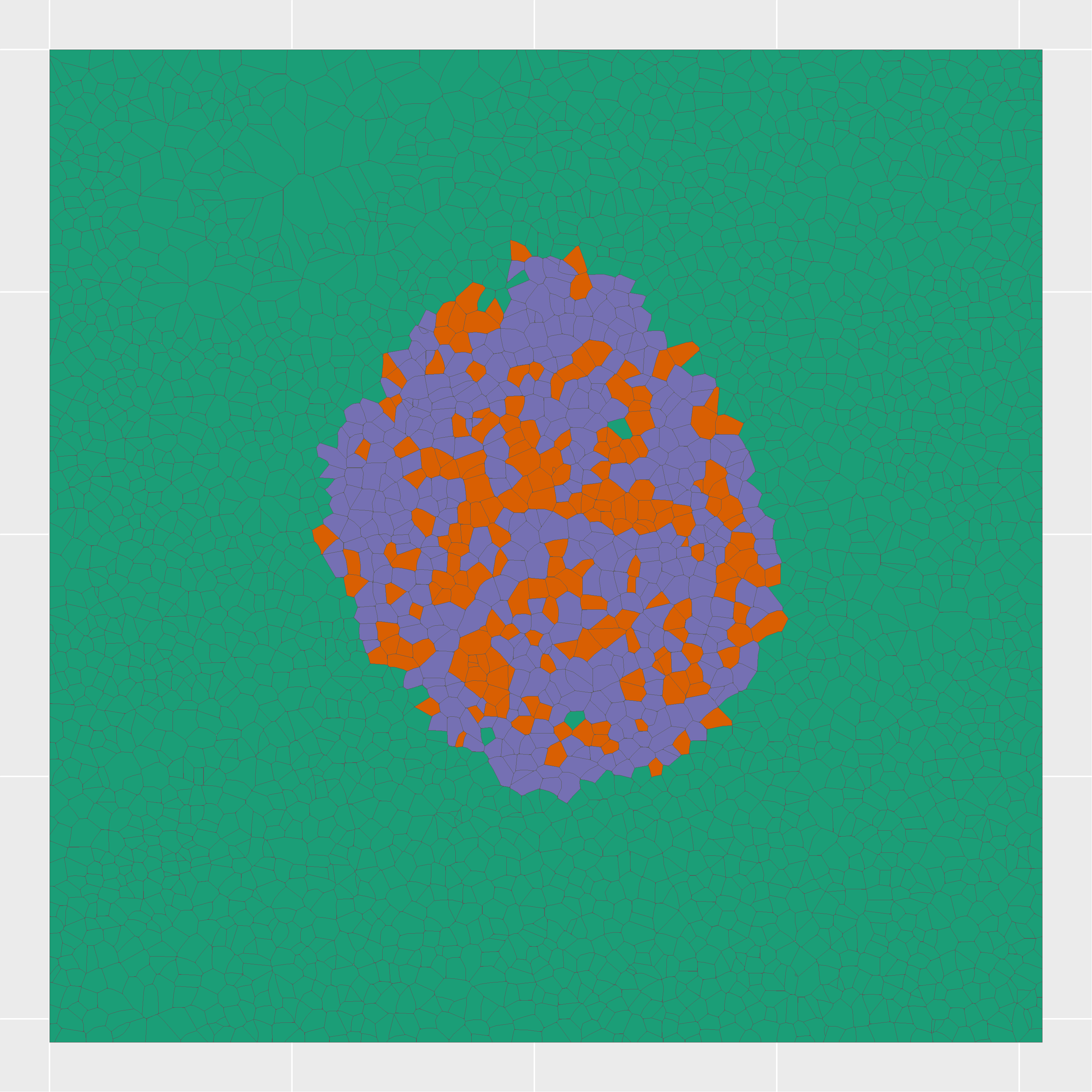
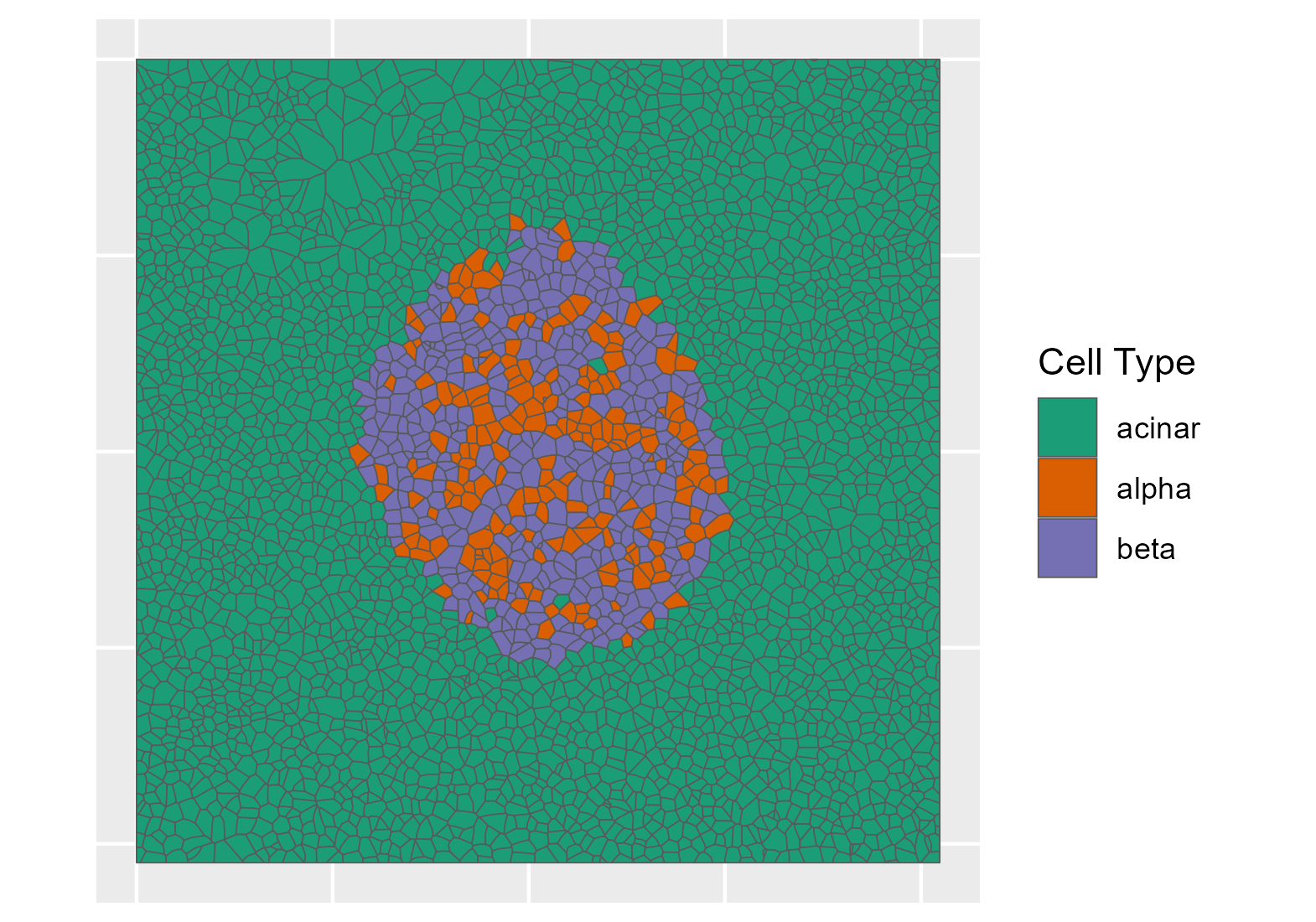
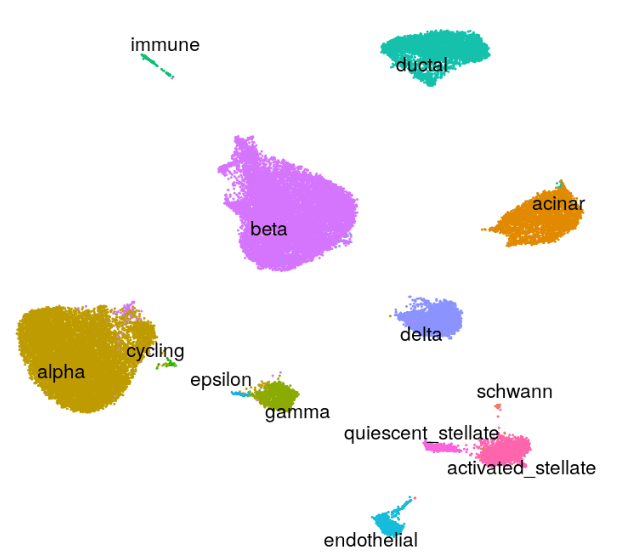
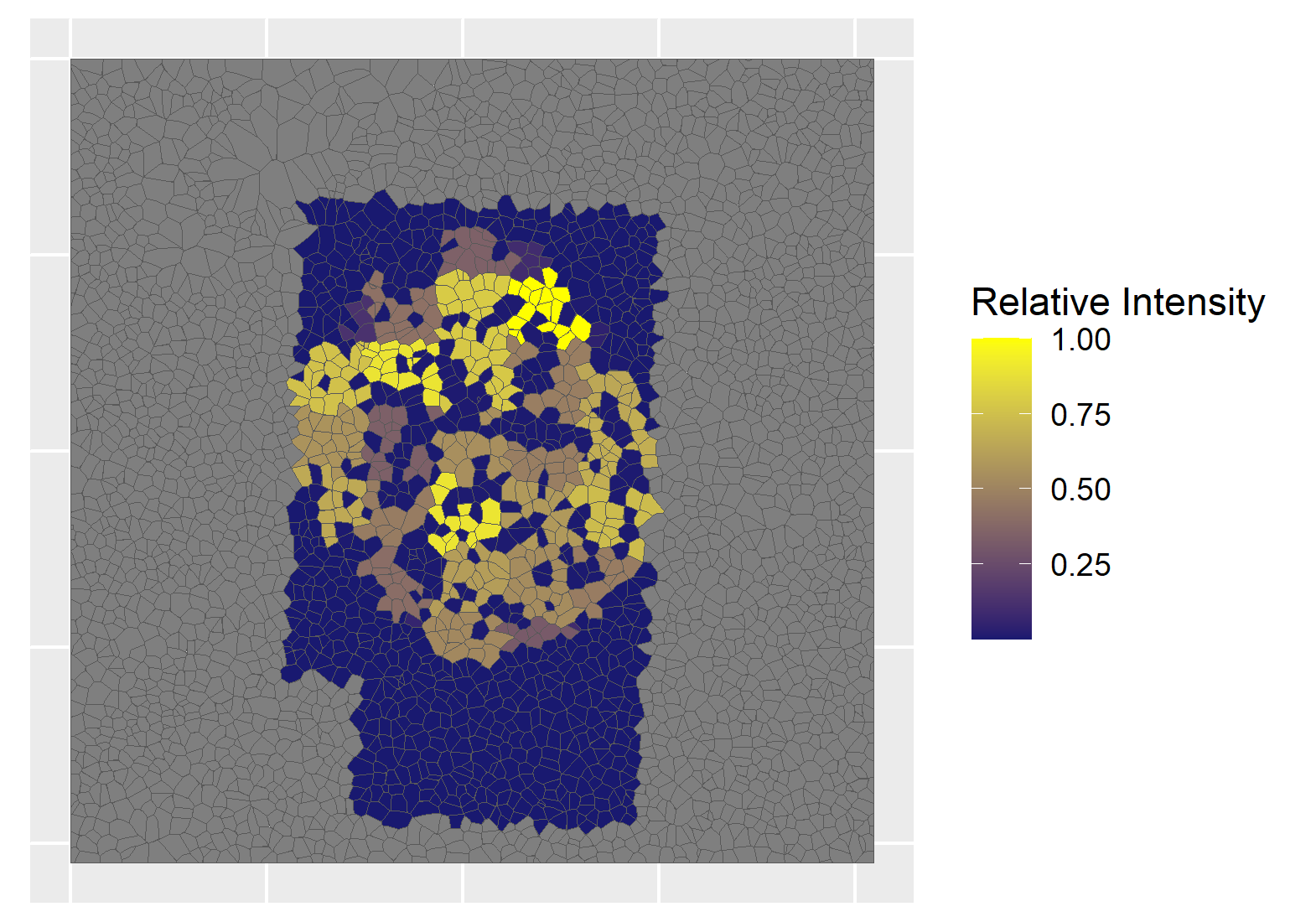
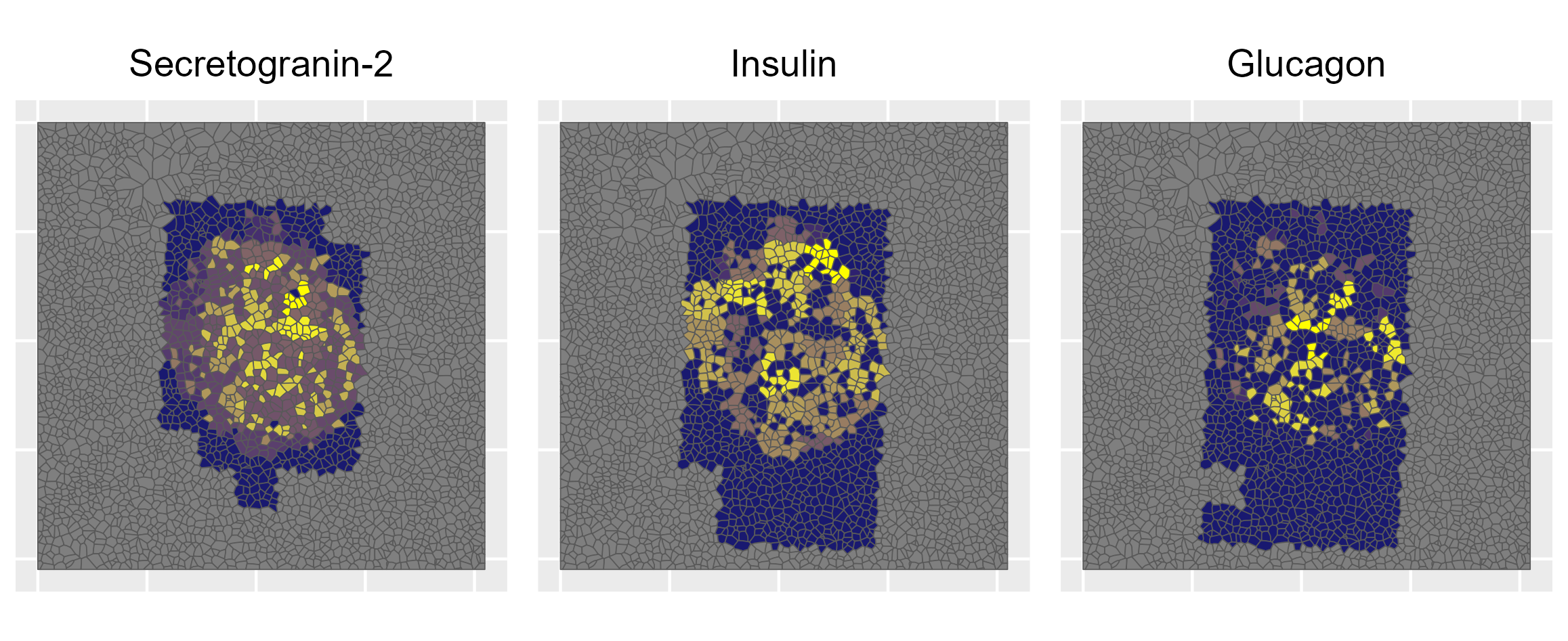
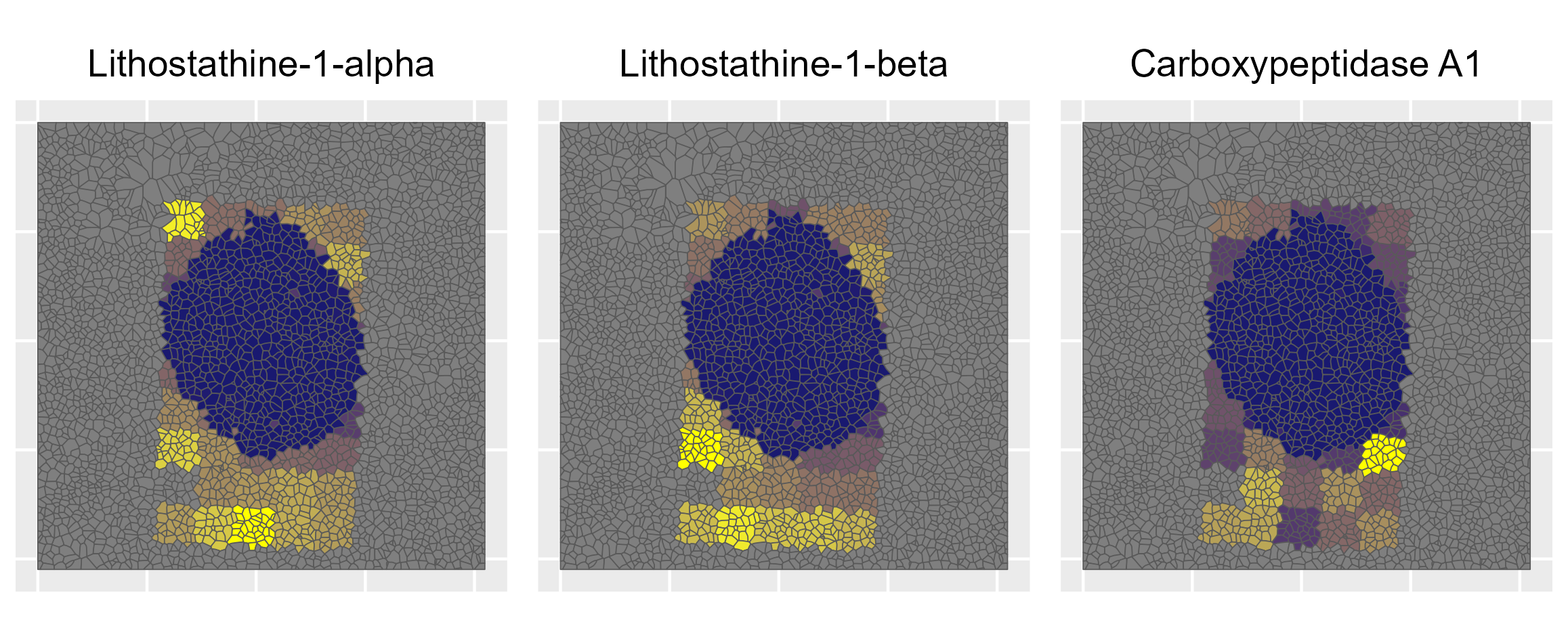
Combining nanopots mass spectrometry with immunofluorescence-guided cell segmentation, we resolve protein abundance across three major cell types—beta (β), alpha (α), and acinar—within a single tissue section.
Cell nuclei were identified with CellPose 2.0 deep learning segmentation, boundaries refined in FIJI, and cell types assigned by INS/GCG/DAPI immunofluorescence channel intensities. Single-cell RNA-seq reference data (Azimuth) was used to deconvolve pixel-level proteomics into cell-type-adjusted protein maps.
2,215Proteins Mapped
3Cell Types Resolved
6Pipeline Stages